|
Looks like the "YLS" have it... Interesting that here we have no functional group name for a compound that is an ester. That would be ester group in my mind. If you look at how a molecular mechanics calculation works and what it reports, "torsional strain" is an increase in energy when four connected atoms are not in their minimum-energy conformation. That's technically not what is stated here, but it's reasonably close. But then at the bottom of page 135 we have only steric strain listed for Me-Me-eclipsed butane. Then is that not also torsional strain? We could perhaps consider the numbers. Eclipsing ethane costs 12 kJ/mol; eclipsing butane costs 16-19 kJ/mol. Seems to me then that most of the strain in eclipsing butane is still torsional strain. I'm not saying Smith says this incorrectly, it's just that it would be nice to explicitly mention that we are talking about a mix of torsional (3-bond) strain and steric (>3-bond) strain. An alternative method of drawing cyclohexane may be found here. See also: Equatorial vs. axial Seeing cis/trans. I don't believe any practicing organic chemist would ever redraw a structure just to figure out R or S when the lowest priority group is in the front. An alternative method of determining R or S when the lowest priority group is in the front is this: Simply determine R or S as always and then just switch the result. Examples of doing that may be found here and here. This problem requires knowledge of alkene isomerization found later in the book, in Chapter 10 on page 363 I prefer the IUPAC recommended name "Gibbs energy" to "Gibbs free energy." More importantly, though, while the notation is correct, the distinction here is not being made between "Gibbs energy" and "standard Gibbs energy." It's a big difference. For EVERY reaction you will ever encounter that actually happens, the change in Gibbs energy will be negative or, at equilibrium, zero. The discussion here refers to standard Gibbs energy, which has the little degree sign with it and does relate as described to the equilibrium constant. This is because
ΔG = ΔGo + RT ln Q
where Q is the reaction quotient, expressed in terms of activities or concentrations. Then, at equilibrium, we have ΔG = 0 and Q = K, resulting in the familiar
ΔGo = - RT ln K Standard Gibbs energy, not just Gibbs energy. Standard Gibbs energy, not just Gibbs energy, same for ΔHo and ΔSo. There isn't much of a difference for ΔH, but there can be a huge difference for ΔS, as that is the primary origin of the reaction quotient:
Sx = Sox - R ln [X]
giving:
ΔS = ΔSo - R ln Q
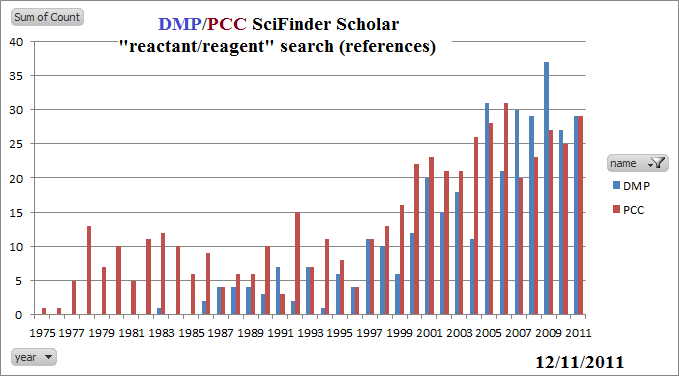
ΔHo arrow is backward; should point downward, from reactants to products, because ΔHo < 0. Example [2]: ΔHo arrow is backward; should point downward, from reactants to products, because ΔHo < 0. Example [3]: ΔHo arrow is backward; should point downward, from reactants to products, because ΔHo < 0. ΔHo arrow is backward; should point downward, from reactants to products, because ΔHo < 0 (in both cases). ΔHo arrow is backward; should point downward, from reactants to products, because ΔHo < 0. ΔHo[2] arrow is backward; should point downward, from reactants to products, because ΔHo < 0. Overall ΔHo arrow is missing; it should point downward. Reactant C- is missing; product A- is missing. This is very important. These are mechanistic steps being described in terms of energy. No energy comparison is possible unless the equation is balanced. Or, to put it another way, comparing the energy of "A-B" to "B-C" is meaningless. Product H2O is missing; this isn't really the mechanism, of course. "HI" does not exist in the presence of water. The words "containing the halogen" should be struck. The longest chain for alkyl halides is determined exactly as for alkanes, because halides, not being able to be represented in the suffix, are not IUPAC principal groups. Just treat fluoro, chloro, bromo, and iodo exactly as you do methyl. It is unnecessary to introduce mechanistic curved arrows at this stage. The discussion is not about mechanism. It is unnecessary to introduce mechanistic curved arrows at this stage. The discussion is not about mechanism. It is unnecessary to introduce mechanistic curved arrows at this stage. The discussion is not about mechanism. NaBr is not soluble in acetone. It's solubility is about 0.001 M [Swearingen, J. Phys Chem 1935, 39, 701-8]. The formation of NaBr as an insoluble product in acetone is the basis for the Finkelstein reaction. A better choice would have been NaI, which is soluble in acetone. ΔHo arrow is backward; should point downward, from reactants to products, because ΔHo < 0. ΔHo[2] and ΔHooverall arrows are backward; should point downward, from reactants to products, because ΔHo < 0. CH3CO2- is missing from reactants; CH3CO2- and Br- are missing from intermediate; Br- is missing from product. There is no such thing as "racemization at a single stereogenic center." And, besides, many SN1 reactions do not result in 50:50 mixtures of isomers. This should read: "SN1 reactions result in a mixture of retention and inversion of stereochemistry at the carbon undergoing the reaction." The equations shown at the top of the page indicate HI and HCl as products. The reactions are done in water. While I suppose these are techincally correct, students need to be thinking about this, and would be helpful to either indicate the products explicitly as H3O+ + I- (or Cl-) or HI(aq)/HCl(aq). The two equations shown in the middle of the page are of great concern to me. These are mechanistic steps -- we are interested in what is really going on here. It is HIGHLY unlikely that Br- would EVER remove the proton from any molecule. In water, HBr, with a pKa of about -9 is fully dissociated. Furthermore, the concentration of Br- relative to water is going to be close to 1: 500. It just won't happen for entropy reasons. For the same reason, HBr cannot be a product of a mechanistic step. The base should be H2O, and the product should be H3O+. DBN and DBU are sterically hindered weak bases, not strong bases. However, they result in Zaytsev-rule elimination, unlike KO-t-Bu. Terminal alkynes cannot be successfully hydroborated using BH3. One needs a dialkylborane such as 9-BBN. BH3 is too reactive and instead reacts twice to form 1,1-diboron compounds. Apparently they do not oxidize well to form aldehydes. The oxidation of alcohols by Cr(VI) is far more complicated than this. It's not clear what is to be gained by suggesting the details shown in this chapter. Green Chemistry -- while it is nice to see a discussion of green chemistry, it seems odd that it should be one that still utilizes Cr(VI) -- one of the most toxic reagents available. The publication of the use of PCC by the Corey group in 1975 was a major advance for organic synthesis, but since then many far less toxic alternatives have been developed. (Saying "Many other green approaches to oxidation that avoid the generation of metal by-products entirely are also under active investigation," though true, seems to miss the point that far greener alternatives to PCC have been available for years and are so well accepted as to no longer be "under active investigation." Surely a far greener alternative would be one of the popular options such as the Swern Oxidation (developed just a few years after PCC and now with over 12,000 references in SciFinder Scholar) or the increasingly popular Dess-Martin Periodinane oxidation. My reading of the synthetic literature is that both PCC and DMP are used in approximately equal frequency as of 2011, but their combined use doesn't come anywhere near that of the Swern oxidation.
Both the Swern oxidation and the Dess-Martin Periodinane reaction have easily understood proposed mechanisms that are relevant to discussions at this level of chemistry. Hooke's law is incorrect. This discussion should include a short introduction to reduced mass. The second bullet should read: The reduced mass (μ) is calculated as
1/μ = 1/m1 + 1/m2
where m1 and m2 are the two masses of the atoms involved in the vibration. The smaller the reduced mass, the higher the energy of the vibration. This explains why bond stretching vibrations for bonds to hydrogen appear so far to the left on the IR spectrum. What is missing from this discussion is the standard way to create a splitting diagram, namely by splitting a peak one H atom at a time. It's a minor point, but I have encountered several students who were totally baffled by this section. This and several other figures in this chapter suffer from what appears to be "artist enhancement" of the actual spectra. This appears as improper secondary effects (heights of the wings of doublets, triplets, and quartets). In this figure, the triplet at δ 3.4 is flipped -- the larger wing should be on the right, not the left. In this figure, the quartet at δ 3.6 is flipped -- the larger wings should be on the right, not the left. Lots of artistic liberty taken here. I'd be interested in knowing what the solvent was that gave that downfield shift to Hb. Possibly d-6 benzene or d-6 DMSO? Whoah! "Carboxylic Acids -- Strong Organic Bronsted-Lowry Acids." I don't think so. Carboxylic acids are weak acids by all measures. I suppose you could argue that "strong organic acids" are "weak acids" and that "strong" here is just a relative term, but my experience is that this is way too subtle a distinction. Further down in this section we also see: "Why are carboxylic acids such strong organic acids? Remember that a strong acid has a weak, stabilized conjugate base. Deprotonation of a carboxylic acid froms a resonance-stabilized conjugate base--a carboxylate anion." So, how do we fix this? The simplest way is just to replace "Strong" in the header with "Weak": "Carboxylic Acids -- Weak Organic Bronsted-Lowry Acids." In the later paragraph, I would suggest: Why are carboxylic acids more acidic than alcohols? Deprotonation of a carboxylic acid froms a resonance-stabilized conjugate base--a carboxylate anion. And leave it at that. What possibly could be gained from showing an energy diagram with three distinct yet identical energies for the three "minor" resonance contributors (2-4) of the phenolate anion? It just seems to me totally inappropriate to do this. Let's reserve energy diagrams for things that can be measured. In any case, even if you could measure this, the species 2-4 would not be at the same energy. I think the real problem starts at the bottom of page 701. The word "stable" doesn't belong in a discussion of resonance contributors, despite the fact that Googling "stable resonance contributor" returns loads of hits. It's not an equilibrium, it's one stable (or not so stable) species. The individual contributors are more or less representative of the actual species, but it is simply not appropriate to talk about their individual "energies" that way.