|
Looks like the "YLS" have it, but you will be right either way. Interesting that here we have no functional group name for a compound that is an ester. That would be carboxylic ester group, I believe. The author herself is confused about what exactly is a compound name and what is a functional group name. Clearly indicated in this figure are circled functional groups. But if you look in Tables 3.1 or 3.2, those are (except for "aromatic ring") explicitly mentioned as types of compounds. Thus, OH is not "alcohol" -- it is a hydroxy (or "hydroxyl") group. The circled nitrogen functional group is not an "amine" -- it is an "amino" group. But if you look in Appendix D, Table D.1, all the names there for "functional groups" are actually names listed in this chapter as "type of compound". Not sure what to make of that. Just sloppy editing, I think. Torsional strain is defined in this text as "an increase in energy caused by eclipsing interactions". While this is a common definition, there are two problems with it. First of all, the word "strain" does not mean "an increase in energy" per se. Strain refers to a deformation from reference positions, both in engineering and in chemistry. If anything, what we have defined here is a "torsional energy". The key is that "torsional strain" is a deformation -- the C-C-H bond angle in ethane is NOT the standard tetrahedral angle of 109.5o in methane or the 111o of ethane. It's more like 112o. So there is strain in this molecule -- a deformation -- and that strain is associated with an increase in energy, which we call tortional energy. But then, later, when we are reading about the strain in butane, we have only steric strain listed for Me-Me-eclipsed butane. My question: Is there not also torsional strain? Of course there is! We could perhaps consider the numbers. Eclipsing ethane costs 12 kJ/mol; eclipsing butane costs 16-19 kJ/mol. Seems to me then that most of the strain in eclipsing butane is still torsional strain. I'm not saying Smith says this incorrectly, it's just that it would be nice to explicitly mention that we are talking about a mix of torsional (3-bond) strain and steric (>3-bond) strain. There are many different ways of drawing the chair conformation of cyclohexane. You will have to figure out what works best for you. An alternative method of drawing cyclohexane may be found here. See also: Equatorial vs. axial Seeing cis/trans. I don't believe any practicing organic chemist would ever redraw a structure just to figure out R or S when the lowest priority group is in the front. It's much easier than that! An alternative method of determining R or S when the lowest priority group is in the front is this: Simply determine R or S as always, only considering the top-three priority groups. Then just switch the result. Examples of doing that may be found here and here. I prefer the IUPAC recommended name "Gibbs energy" to "Gibbs free energy." More importantly, though, while the notation is correct, the distinction here is not being made between "Gibbs energy" and "standard Gibbs energy." It's a big difference. For EVERY reaction you will ever encounter that actually happens, the change in Gibbs energy will be negative or, at equilibrium, zero. The discussion here refers to the standard Gibbs energy of reaction, which has a delta before it and the little degree sign with it (as shown correctly here) and does relate as described to the equilibrium constant. This is because
ΔG = ΔGo + RT ln Q
where Q is the reaction quotient, expressed in terms of activities or concentrations. Then, at equilibrium, we have ΔG = 0 and Q = K, resulting in the familiar
ΔGo = - RT ln K
So while I appreciate the use of the superscript o, it is not proper to call this Gibbs energy or Gibbs free energy. It is the standard Gibbs (free) energy. Standard Gibbs energy change, not just Gibbs energy change, same for ΔHo (standard enthalpy change) and ΔSo (standard entropy change). There isn't much of a difference for ΔH, but there can be a huge difference for ΔS, as that is the primary origin of the reaction quotient:
Sx = Sox - R ln [X]
giving:
ΔS = ΔSo - R ln Q
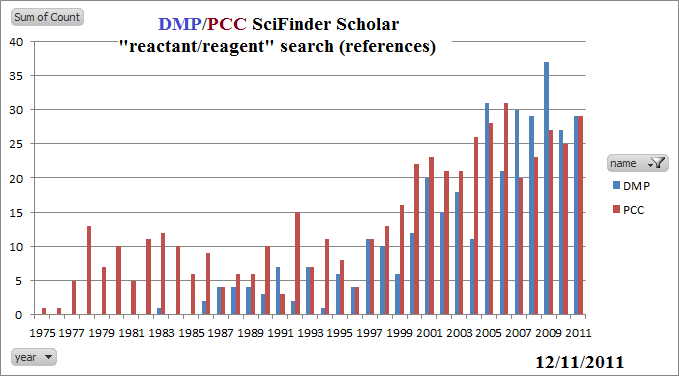
This is really important. I simply do not understand why the editors of this book, after six editions have not corrected these diagrams. The problem appears over and over: The ΔHo arrow is backward; it should point downward, from reactants to products, because ΔHo < 0 and, well, because just like a reaction arrow, this arrow should always go from reactant to product. What is so hard about that!? Upward pointing arrows are positive changes, downward negative, in all of mathematics and chemistry. All I can guess is that the artist made a mistake, the editors did not do their jobs, and, apparently, McGraw-Hill is too cheap to correct the problem even after being told multiple times that it is a simple fix. Example [2]: ΔHo arrow is backward; should point downward, from reactants to products, because ΔHo < 0. Example [4]: ΔHo arrow is backward; should point downward, from reactants to products, because ΔHo < 0. ΔHo arrow is backward; should point downward, from reactants to products, because ΔHo < 0. ΔHo[2] arrow is backward; should point downward, from reactants to products, because ΔHo < 0. Overall ΔHo arrow is missing; it should point downward. The "overall" standard enthalpy change is negative; its arrow, were it there, should go down. There are just so many things wrong with this figure. A key principle of chemistry is that reaction equations must be balanced. This applies jsut as well to reaction diagrams. This diagram states that "A-B ⟶ B-C", which is nonsense. Reactant C- is missing; product A- is missing. This is sloppy editing and is very important. These are mechanistic steps being described in terms of energy. No energy comparison is possible unless the equation is balanced. Or, to put it another way, comparing the energy of "A-B" to "B-C" is meaningless. The phrase , with the halogen as a substituent of the longest chain should be struck. The longest chain for alkyl halides is determined exactly as for alkanes, without regard to whether or not there is a halogen, because halo groups, not being able to be represented in the suffix, are not IUPAC principal groups. Just treat fluoro, chloro, bromo, and iodo exactly as you do methyl. NaBr is not soluble in acetone. It's solubility is about 0.001 M [Swearingen, J. Phys Chem 1935, 39, 701-8]. The formation of NaBr as an insoluble product in acetone is the basis for the Finkelstein reaction. The author is probably thinking of sodium iodide, which is (amazingly) soluble in acetone. ΔHo arrow is backward; should point downward, from reactants to products, because ΔHo < 0. ΔHo[2] and ΔHooverall arrows are backward; should point downward, from reactants to products, because ΔHo < 0. All equations in chemistry need to be charge and mass balanced. Neither is the case here. CH3CO2- is missing from reactants; CH3CO2- and Br- are missing from intermediate; Br- is missing from product. It is not possible to compare the energies of an alkyl bromide with an alkyl acetate, nor either of these with the energy of a cation. This makes no sense. There is no such thing as "racemization at a single stereogenic center." Racemization is a process of producing a racemic mixture of compounds. And, besides, many SN1 reactions do not result in 50:50 mixtures of isomers. It is a gross simplification and provably false that "SN1 reactions proceed with racemization at a single stereogenic center." This is never the case, for example, when the compound reacting has two stereocenters, one of which is unreactive. This should read: "SN1 reactions result in a mixture of retention and inversion of stereochemistry at the carbon undergoing the reaction." The equations shown indicate HI and HCl as products. But we know this cannot be the case. The reactions are done in water, where both HI and HCl are strong acids. While I suppose these are techincally correct, students need to be thinking about this, and would be helpful to either indicate the products explicitly as H3O+ + I- (or Cl-) or HI(aq)/HCl(aq). The two equations shown in the middle of the page are not acceptable mechanistic steps. They are shown as bimolecular steps involving the extremely weak base Br-. However, this is simply not possible (or, I should say, extremely unlikely). There is a reason we call HBr a strong acid, after all. It is fully dissociated in water. So what is pulling off the proton in this step? (It is certainly not just falling off on its own.) Well, what is in this solution? Bromide and water, right? Which is more basic? (What is the pKa of their conjugate acids?) Furthermore, the concentration of water relative to Br- is going to be close to 500:1. The base should be H2O, and the product should be H3O+ in both cases. The Br- is just a spectator ion (surrounded by a shell of associated water molecules) once the alkyl halide dissociates. For the same reason, HBr cannot possibly be a product of a mechanistic step, either. I'm sorry, but DBN and DBU are sterically hindered, nonnucleophilic weak bases, not strong bases. (That said, with a pKa around 12.5, they are about 100 times stronger than triethylamine. Still, this means they are about 10,000 times weaker than hydroxide.) Note that they result in Zaytsev-rule elimination, unlike KO-t-Bu. The oxidation of alcohols by Cr(VI) is far more complicated than this. What is shown here has no basis in fact whatsoever. It's not clear what is to be gained by suggesting the details shown in this chapter. Green Chemistry -- while it is nice to see a discussion of green chemistry, it seems odd that it should be one that still utilizes Cr(VI) -- one of the most toxic reagents available. The publication of the use of PCC by the Corey group in 1975 was a major advance for organic synthesis, but since then many far less toxic alternatives have been developed. Saying "Many other green approaches to oxidation that avoid the generation of metal by-products entirely are also under active investigation," though true (throughout 4 editions, though??), seems to miss the point that far greener alternatives to PCC have been available for years and are so well accepted as to no longer be "under active investigation." For example, a far greener alternative would be one of the popular options such as the Swern Oxidation (developed just a few years after PCC and now with over 12,000 references in SciFinder Scholar) or the increasingly popular Dess-Martin Periodinane oxidation. My reading of the synthetic literature is that both PCC and DMP are used in approximately equal frequency as of 2011, but their combined use doesn't come anywhere near that of the Swern oxidation.
Both the Swern oxidation and the Dess-Martin Periodinane reaction have easily understood proposed mechanisms that are relevant to discussions at this level of chemistry. Hooke's law is incorrect. This discussion should include a short introduction to reduced mass. The second bullet should read: The reduced mass (μ) is calculated as
1/μ = 1/m1 + 1/m2
where m1 and m2 are the two masses of the atoms involved in the vibration. The smaller the reduced mass, the higher the energy of the vibration. This explains why bond stretching vibrations for bonds to hydrogen appear so far to the left on the IR spectrum. This and several other figures in this chapter suffer from what appears to be "artist enhancement" of the actual spectra. This appears as improper secondary effects (heights of the wings of doublets, triplets, and quartets). In this figure, the triplet at δ 3.4 is flipped -- the larger wing should be on the right, not the left. The statement in this section, "Imine formation is most rapid at pH 4-5." is incorrect. Apparently, your author picked this statement up from a superficial reading of another textbook. Here is a message on the Chemistry Education Listserve from May 12, 2015:
Hopefully this will answer your questions about imine formation: The pKa value of a protonated carbonyl group of an aldehyde or ketone is about -8 to -7. The pKa value of a protonated primary amine is about 11. Therefore, the protonated carbonyl compound is about 10 to the 19th times more acidic than a protonated amine, so it is impossible to have any protonated carbonyl compound in a solution that contains any unprotonated amine. Imine formation should be carried out about 1.5 pH units lower that the pKa of the primary ammonium ion----and since primary ammonium ions have pKa values of about 11, imine formation should be carried out at about pH 9.5. (At pH 9.5 there is some nonprotonated amine in solution but there is NO protonated carbonyl compound.) Therefore, in the first step of the reaction, the negative charge is not being formed in an acidic solution. I am very sorry that so many people now think that imine formation should be carried out at pH 4-5, because I think the origin of this error results from people misreading my textbook. My textbook has a pH rate-profile for imine formation that shows a maximum value at pH 4.5. That pH-rate profile comes from a JACS paper by Bill Jencks. The amine he used to generate the pH-rate profile was hydroxylamine whose ammonium ion has a pKa value of 6.0. Therefore, pH 4.5 is 1.5 units lower that the pKa of the amine. It would have been impossible to obtain a pH rate-profile in the pH range needed if he had used a protonated ammonium ion with at pKa of 11, thus his decision to use hydroxylamine and the reason people now think that imine formation should be carried out at pH 4.5. The Mechanism The first step is amine addition to the neutral ketone or aldehyde to form a tetrahedral intermediate (TI) with negatively charged oxygen that then is protonated. The next step is to lose a proton from the N-protonated TI to form a neutral TI. The neutral TI can be re-protonated on N or protonated on O. Re-protonation on N pushes the equilibrium back to starting materials. Protonation on O leads to loss of water and iminium ion formation. Protonation on N is favored because N is a stronger base than O. That is why imine formation requires water to be removed as the imine is formed----because of the preference to protonate N rather than O. I hope this helps. Paula Paula Yurkanis Bruice, Ph.D. Department of Chemistry and Biochemistry University of California, Santa Barbara, CA 93106Whoah! "Carboxylic Acids -- Strong Organic Bronsted-Lowry Acids." I don't think so. Carboxylic acids are weak acids by all measures. I suppose you could argue that "strong organic acids" are "weak acids" and that "strong" here is just a relative term, but my experience is that this is way too subtle a distinction. Further down in this section we also see: "Why are carboxylic acids such strong organic acids? Remember that a strong acid has a weak, stabilized conjugate base. Deprotonation of a carboxylic acid froms a resonance-stabilized conjugate base--a carboxylate anion." So, how do we fix this? The simplest way is just to replace "Strong" in the header with "Weak": "Carboxylic Acids -- Weak Organic Bronsted-Lowry Acids." In the later paragraph, I would suggest: Why are carboxylic acids more acidic than alcohols? Deprotonation of a carboxylic acid froms a resonance-stabilized conjugate base--a carboxylate anion. And leave it at that. This "energy diagram" is meaningless and a total fabrication from imagination. Resonance contributers cannot be compared energetically. They can be compared in terms of their relative weight in representing the "true" character of a species, but that is all. No numbers could ever be assigned to these "states". There is no equilibrium; these different "states" do not exist independently. Let's reserve energy diagrams for things that can be measured. In any case, even if you could measure this, the species 2, 3, and 4 would not be at the same energy. I think the real problem starts just above thiss figure. The word "stable" doesn't belong in any discussion of resonance contributors, despite the fact that Googling "stable resonance contributor" returns loads of hits. It's not an equilibrium, it's one stable (or not so stable) species. The individual contributors are more or less representative of the actual species, but it is simply not appropriate to talk about their individual "energies" of "resonance structures" that way.